如何发现宏基因组研究中的偏见
宏基因组研究的增长彻底改变了我们对微生物群与环境或健康之间关系的理解。
尽管这种认识导致了许多新发现,但数据可重复性仍然是一个挑战。该问题涵盖了跨实验室的宏基因组研究,源于以下事实:偏见可以在宏基因组工作流程的各个步骤中引入偏见,如该领域的许多人所观察到的那样1-6。
偏见的问题是如此普遍,即使将相同的样本提交给两个不同的微生物组分析组织也可以产生彼此之间截然不同的结果(图1)。
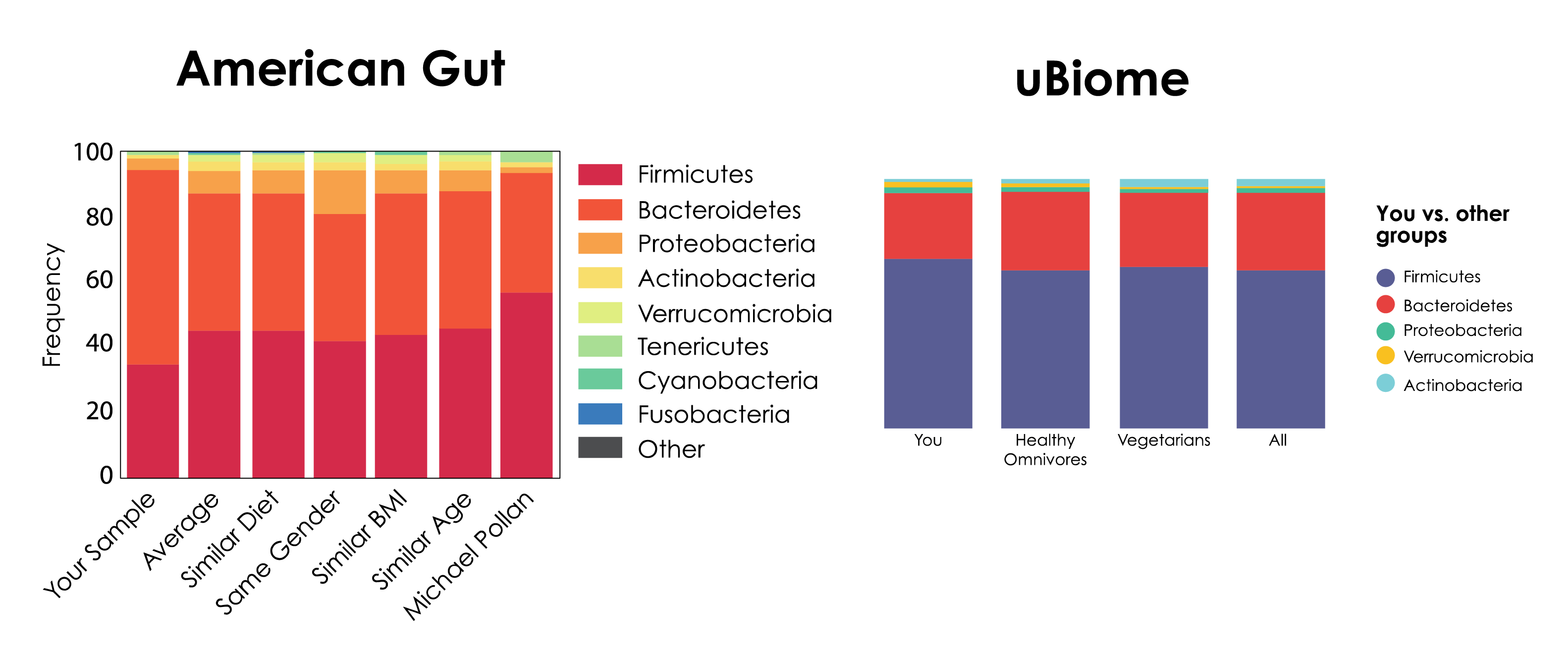
这些偏差可能会在整个宏基因组学工作流程中的每一步都产生。但是,导致偏见的最有问题的步骤之一在于核酸提取。有了越来越多的全身偏见的证据,现在比以往任何时候都大得多准确的宏基因组核酸提取工作流程。
提取中如何发生偏见?
微生物群落是复杂而多样的,包括革兰氏阳性细菌,革兰氏阴性细菌和真菌。准确的元素分析需要从微生物群落中所有不同物种中解放DNA。然而,通常观察到核酸提取过程中无效的裂解,从而导致微生物剖面偏置。这是由于某些微生物很难裂解6,8。如果细胞未裂解,DNA将保持锁定在细胞内,并且不会被纯化或检测到。
已经表明,由于这种原因,使用化学或热裂解过度占用易于散热的生物(革兰氏阴性细菌)的过程。由于很难进行的生物(例如革兰氏阳性细菌和酵母)对DNA释放具有更耐药性,因此它会对易于散热物种产生偏见。许多提取方案在样本组成中没有考虑到这些巨大差异,这意味着观察到不均匀裂解和微生物谱偏见是常见的9。
利用机械裂解的提取方案(例如超声处理,液氮/砂浆和杵,法式压迫和珠子)被认为是微生物裂解的最佳方法由于其随机性质,珠子跳动被称为黄金标准。但是,这些机械裂解方法仍然需要优化,或者它们将遭受低产量,过度核酸剪切,非均匀裂解,过量热和剪切力等问题。
如何发现偏见?
唯一知道提取系统是否将偏见引入宏基因组研究的唯一真实方法是评估该系统微生物标准。微生物标准是指各种微生物(包括革兰氏阳性和革兰氏阴性物种),它们充当模拟微生物群落,并模仿样品中存在的宏基因组种群。该标准通过提取工作流程正常处理。
由于已知每种微生物在微生物标准中的丰度,因此从16S测序数据获得的结果应与标准匹配。与此的大偏差表明,提取系统将偏置引入结果。最常见的是,这些偏差表明自己是人口中革兰氏阴性物种的过度占代表性。在对各种提取系统的比较中可以清楚地看到这一点(图2)。
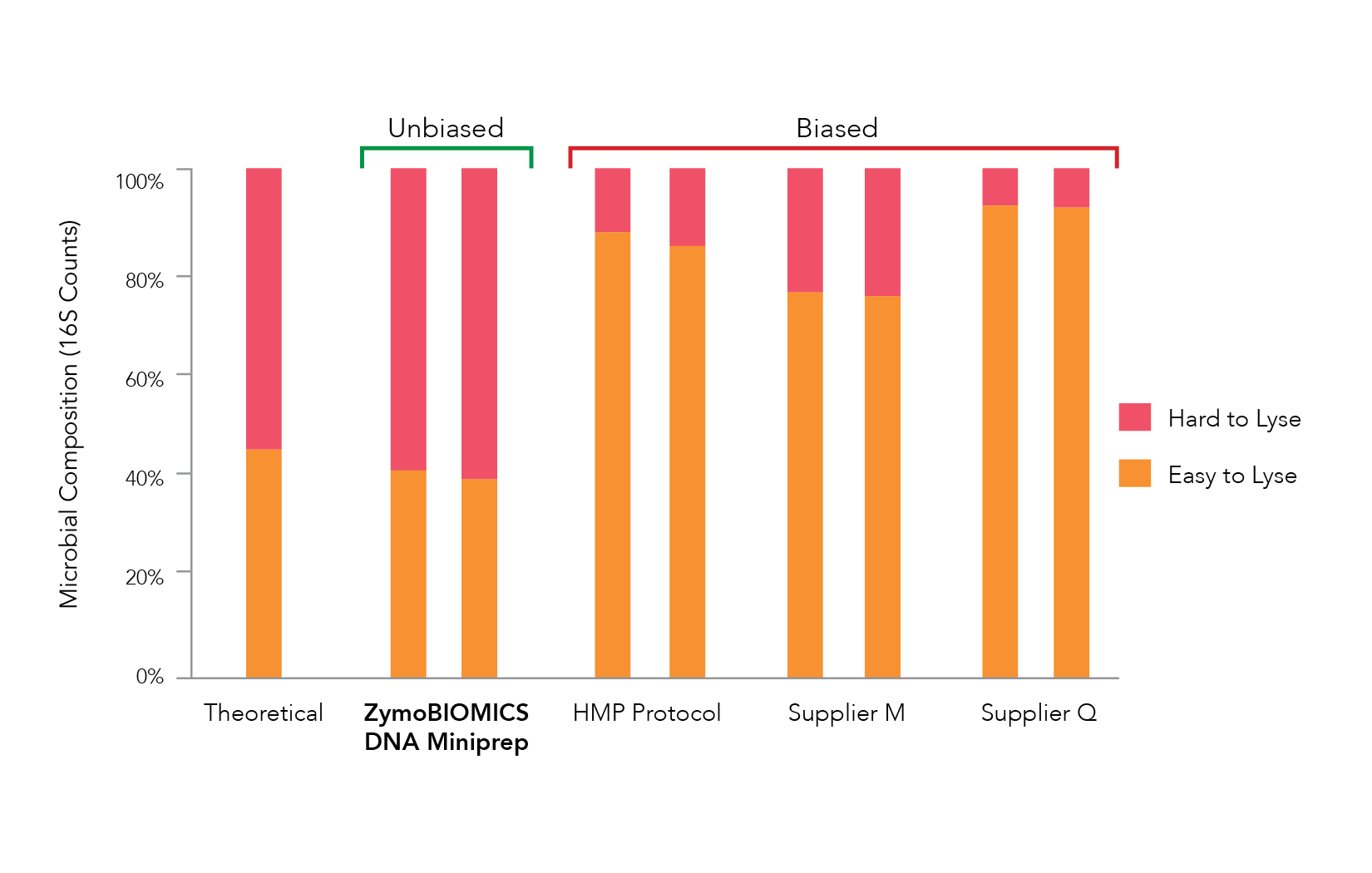
无偏见的方法
Zymobiomics系列解决了宏基因组工作流程中偏差的关键挑战。这Zymobiomics 96 Magbead DNA套件利用已通过微生物社区标准进行开发和优化的机械裂解,以确保对所有难以散热的生物的完全裂解(图3)。
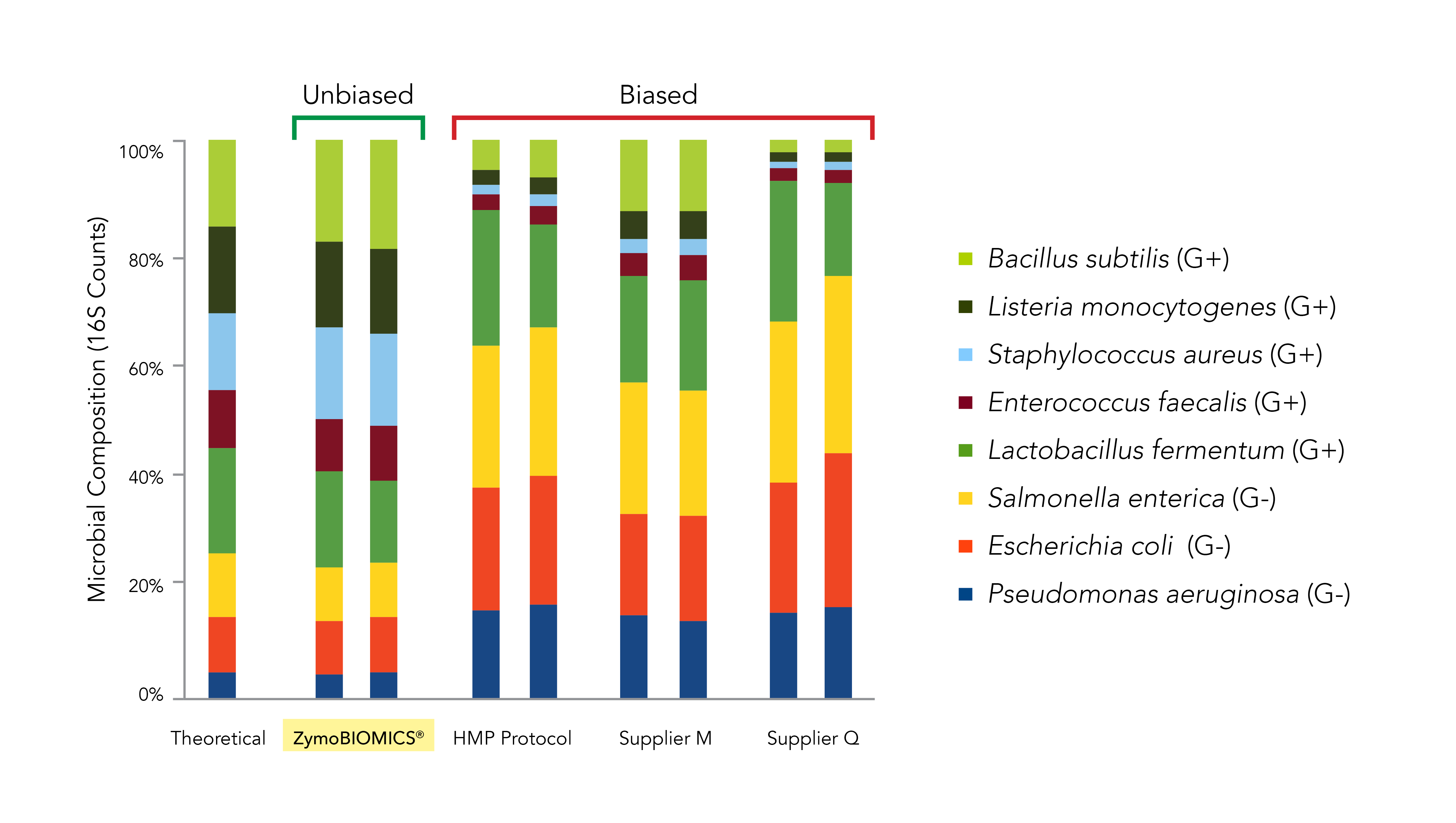
- Sinha R,Abnet CC,White O,Knight R,Huttenhower C:微生物组质量控制项目:基线研究设计和未来方向。Genome Biol 2015,16:276。
- Hsieh YH,Peterson CM,Raggio A,Keenan MJ,Martin RJ,Ravussin E,Marco ML:不同粪便加工方法对人类肠道细菌多样性评估的影响。微生物学的前沿2016,7:1643。13。
- Vishnivetskaya TA,Layton AC,Lau MC,Chauhan A,Cheng KR,Meyers AJ,Murphy JR,Rogers AW,Saarunya GS,Williams de等人:商业DNA提取套件影响了Permafrost Samples中观察到的微生物群落组成。FEMS微生物学生态学2014,87(1):217-230。14。
- Hart ML,Meyer A,Johnson PJ,Ericsson AC:从多个宿主物种的粪便中进行DNA提取方法的比较评估,用于下游下一代测序。PLOS ONE 2015,10(11):E0143334。15。
- Kennedy NA,Walker AW,Berry SH,Duncan SH,Farquarson FM,Louis P,Thomson JM,Satsangi J,Flint HJ,Parkhill J等:不同DNA提取试剂盒和实验室对人类肠道肠道微生物群的影响,由人类的guut Microbiota组成。16S rRNA基因测序。PLOS ONE 2014,9(2):E88982。16。
- Sohrabi M,Nair RG,Samaranayake LP,Zhang L,Zulfiker AH,Ahmetagic A,Good D,Wei MQ:来自人类口服RINSE样品的细胞和细菌DNA提取物的产率和质量受到细胞裂解方法的变量影响。微生物方法杂志2016,122:64-72。
- Saey TH:这是对您的肠道微生物组进行分析的大便。在:科学新闻。卷。2017;2014。
- FarkašV,Takeo K,MacekováD,Ohkusu M,Yoshida S,Sipiczki M.加密型新羊角菌中的次要细胞壁形成是一种反对酸诱导自动解的救援机制。FEMS酵母研究,2009,9(2):311-320
- Costea等。迈向宏基因组研究中人类粪便样品加工标准。自然生物技术(2017)11:1069-1076





