Microbiomics: Macro-Bias
测量领域的领导人不同意…
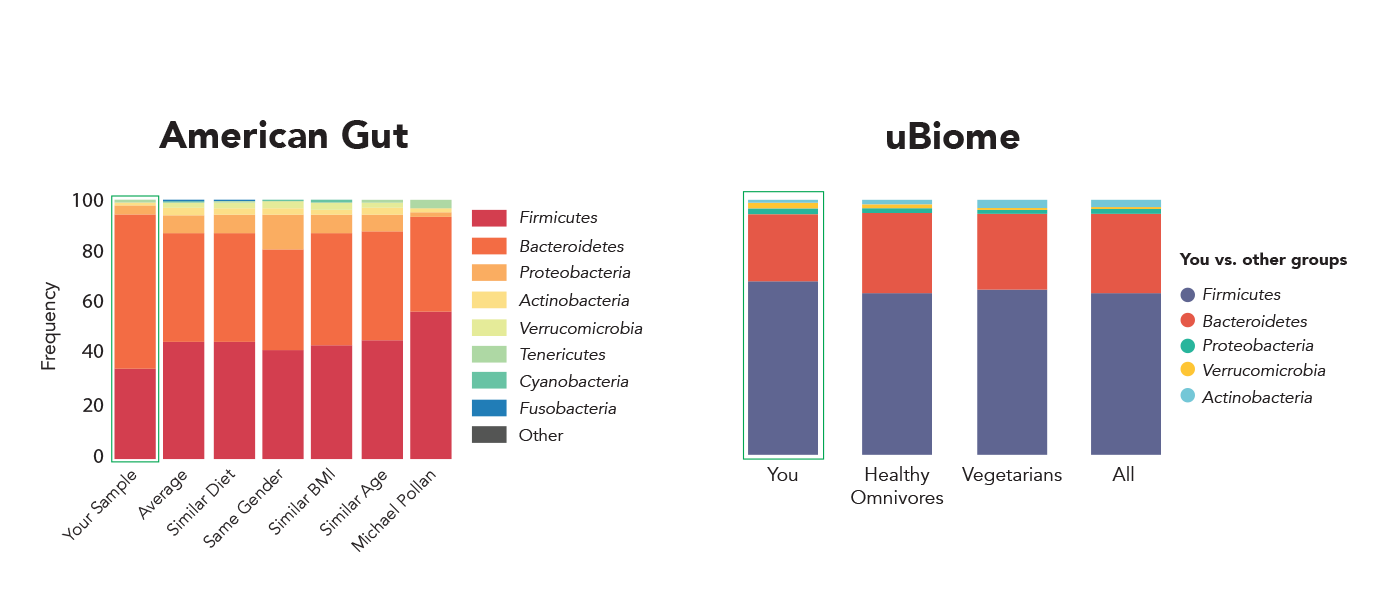
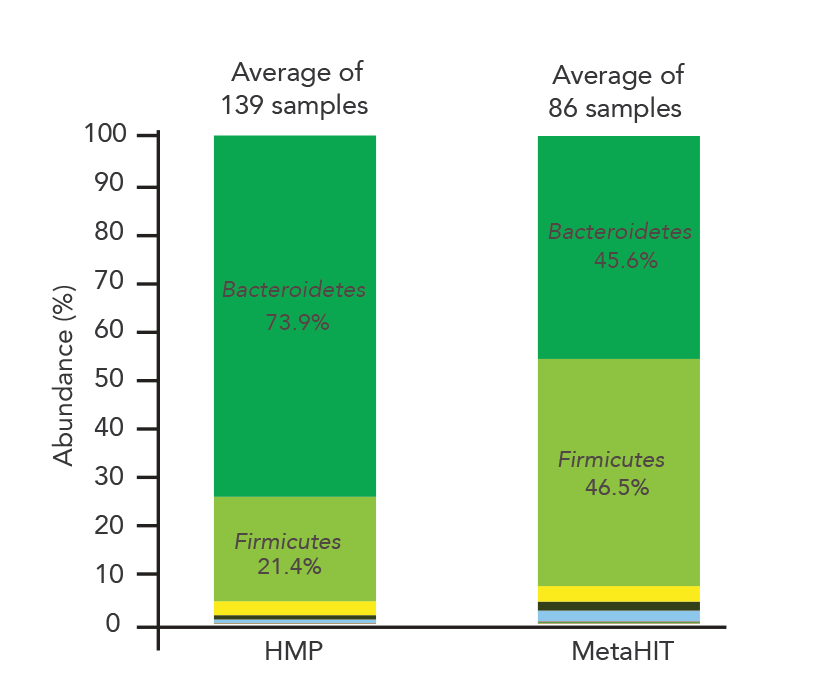
如果你发送相同的样本到两个不同的实验室进行分析,同意你所期望的结果。不幸的是,这还没有微生物组测量甚至来自著名的组织。美国肠道和uBiome是两个最著名的微生物分析组织。但是,给定相同的粪便样本,两个组织报道截然不同的微生物概要文件(图1)[1]。甚至更突出的,最大的两个微生物分析项目,人类微生物组计划(HMP)和宏基因组的人类肠道(MetaHIT),分析了成百上千的人类肠道微生物样品,发现两个主要类群(拟杆菌门和壁厚菌门)截然不同的丰度(图2)。不幸的是,这种差异可能是由于技术变化在DNA提取程序所使用的两个项目是Wesolowska-Andersen et al。[2]指出。
会出现什么问题呢?
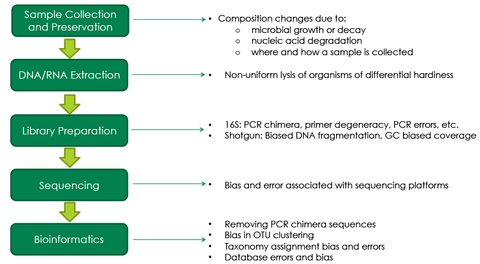
上述矛盾的结果最有可能的是由于偏见产生由于微生物的复杂性质测量。一个门店microbiomics工作流有多个步骤,每个步骤都有各种因素,可以因为偏见最终结果(图3)。之前microbiomics实验设计中使用的大多数协议目前microbiomics领域的发展;因此,他们没有设计错综复杂的偏见。例如,尽管传统的DNA提取方法生成的高度纯净的DNA,它可能只提取DNA只从easy-to-lyse细菌而留下tough-to-lyse细菌DNA导致明显不准确的资料。
可以做些什么?
微生物组测量要求更严格的认证无偏和较低的微生物污染水平的方法。帮助研究人员产生准确的微生物组测量,Zymo研究领先新Microbiome-Grade工具这一领域的发展。这些包括第一个商用微生物参考资料,第一个DNA提取工具旨在确保无细胞裂解,和第一cold-free和公正的方法保护微生物样品。
了解更多关于Microbiome-Grade工具集的标准准确微生物分析:
引用:
1。Saey TH:这是粪便在肠道微生物分析。:科学新闻。卷。2017;2014年。
2。Wesolowska-Andersen,巴尔MI,卡瓦略V, Kristiansen K, Sicheritz-Ponten T,古普塔R,轻轻地TR:从粪便细菌的DNA提取方法的选择影响群落结构评估的宏基因组分析。微生物组2014,19。





